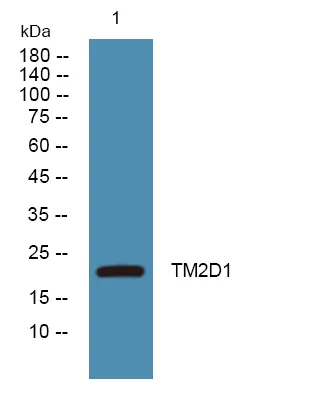
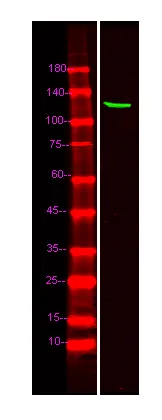
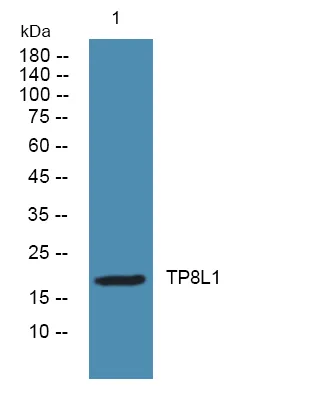
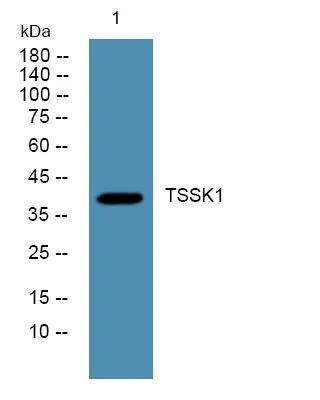
产品概述
产品性能
免疫原
产品应用
研究背景
disease:Defects in MT-ATP6 are a cause of infantile bilateral striatal necrosis [MIM:500003]. Bilateral striatal necrosis is a neurological disorder resembling Leigh syndrome.,disease:Defects in MT-ATP6 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-ATP6 are a cause of Leigh syndrome (LS) [MIM:256000]. LS is a severe neurological disorder characterized by bilaterally symmetrical necrotic lesions in subcortical brain regions.,disease:Defects in MT-ATP6 are the cause of neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP) [MIM:551500].,disease:Defects in MT-CO3 are a cause of cytochrome c oxidase deficiency (COX deficiency) [MIM:220110]; also called mitochondrial complex IV deficiency. COX deficiency is a clinically heterogeneous disorder. The clinical features are ranging from isolated myopathy to severe multisystem disease, with onset from infancy to adulthood.,disease:Defects in MT-CO3 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-CO3 are associated with recurrent myoglobinuria [MIM:550500]. Myoglobinuria consists of excretion of myoglobin in the urine.,disease:Defects in MT-CO3 are found in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome, a genetically heterogeneous disorder, characterized by episodic vomiting, seizures, and recurrent cerebral insults resembling strokes and causing hemiparesis, hemianopsia, or cortical blindness.,function:Mitochondrial membrane ATP synthase (F(1)F(0) ATP synthase or Complex V) produces ATP from ADP in the presence of a proton gradient across the membrane which is generated by electron transport complexes of the respiratory chain. F-type ATPases consist of two structural domains, F(1) - containing the extramembraneous catalytic core and F(0) - containing the membrane proton channel, linked together by a central stalk and a peripheral stalk. During catalysis, ATP synthesis in the catalytic domain of F(1) is coupled via a rotary mechanism of the central stalk subunits to proton translocation. Key component of the proton channel; it may play a direct role in the translocation of protons across the membrane.,function:Mitochondrial membrane ATP synthase (F(1)F(0) ATP synthase or Complex V) produces ATP from ADP in the presence of a proton gradient across the membrane which is generated by electron transport complexes of the respiratory chain. F-type ATPases consist of two structural domains, F(1) - containing the extramembraneous catalytic core and F(0) - containing the membrane proton channel, linked together by a central stalk and a peripheral stalk. During catalysis, ATP synthesis in the catalytic domain of F(1) is coupled via a rotary mechanism of the central stalk subunits to proton translocation. Part of the complex F(0) domain. Minor subunit located with subunit a in the membrane.,function:Subunits I, II and III form the functional core of the enzyme complex.,similarity:Belongs to the ATPase A chain family.,similarity:Belongs to the ATPase protein 8 family.,similarity:Belongs to the cytochrome c oxidase subunit 3 family.,subunit:F-type ATPases have 2 components, CF(1) - the catalytic core - and CF(0) - the membrane proton channel.,subunit:F-type ATPases have 2 components, CF(1) - the catalytic core - and CF(0) - the membrane proton channel. CF(1) has five subunits: alpha(3), beta(3), gamma(1), delta(1), epsilon(1). CF(0) has three main subunits: a, b and c.,disease:Defects in MT-ATP6 are a cause of infantile bilateral striatal necrosis [MIM:500003]. Bilateral striatal necrosis is a neurological disorder resembling Leigh syndrome.,disease:Defects in MT-ATP6 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-ATP6 are a cause of Leigh syndrome (LS) [MIM:256000]. LS is a severe neurological disorder characterized by bilaterally symmetrical necrotic lesions in subcortical brain regions.,disease:Defects in MT-ATP6 are the cause of neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP) [MIM:551500].,disease:Defects in MT-CO3 are a cause of cytochrome c oxidase deficiency (COX deficiency) [MIM:220110]; also called mitochondrial complex IV deficiency. COX deficiency is a clinically heterogeneous disorder. The clinical features are ranging from isolated myopathy to severe multisystem disease, with onset from infancy to adulthood.,disease:Defects in MT-CO3 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-CO3 are associated with recurrent myoglobinuria [MIM:550500]. Myoglobinuria consists of excretion of myoglobin in the urine.,disease:Defects in MT-CO3 are found in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome, a genetically heterogeneous disorder, characterized by episodic vomiting, seizures, and recurrent cerebral insults resembling strokes and causing hemiparesis, hemianopsia, or cortical blindness.,function:Mitochondrial membrane ATP synthase (F(1)F(0) ATP synthase or Complex V) produces ATP from ADP in the presence of a proton gradient across the membrane which is generated by electron transport complexes of the respiratory chain. F-type ATPases consist of two structural domains, F(1) - containing the extramembraneous catalytic core and F(0) - containing the membrane proton channel, linked together by a central stalk and a peripheral stalk. During catalysis, ATP synthesis in the catalytic domain of F(1) is coupled via a rotary mechanism of the central stalk subunits to proton translocation. Key component of the proton channel; it may play a direct role in the translocation of protons across the membrane.,function:Mitochondrial membrane ATP synthase (F(1)F(0) ATP synthase or Complex V) produces ATP from ADP in the presence of a proton gradient across the membrane which is generated by electron transport complexes of the respiratory chain. F-type ATPases consist of two structural domains, F(1) - containing the extramembraneous catalytic core and F(0) - containing the membrane proton channel, linked together by a central stalk and a peripheral stalk. During catalysis, ATP synthesis in the catalytic domain of F(1) is coupled via a rotary mechanism of the central stalk subunits to proton translocation. Part of the complex F(0) domain. Minor subunit located with subunit a in the membrane.,function:Subunits I, II and III form the functional core of the enzyme complex.,similarity:Belongs to the ATPase A chain family.,similarity:Belongs to the ATPase protein 8 family.,similarity:Belongs to the cytochrome c oxidase subunit 3 family.,subunit:F-type ATPases have 2 components, CF(1) - the catalytic core - and CF(0) - the membrane proton channel.,subunit:F-type ATPases have 2 components, CF(1) - the catalytic core - and CF(0) - the membrane proton channel. CF(1) has five subunits: alpha(3), beta(3), gamma(1), delta(1), epsilon(1). CF(0) has three main subunits: a, b and c.,
研究领域